
NovaSeq™ X Plus是因美纳史上通量最大、产能最高、可持续性最强的高通量测序仪,每年可以测序超过20,000个人类标准全基因组,能够支持更大规模的测序、更多种类的研究;其中全新的XLEAP-SBS化学技术能带来更快速度、更优质量、更高可靠性。诺禾致源天津实验室现已完成NovaSeq X Plus全基因组、全外显子组、转录组、Meta文库测序数据分析。本次测评从测序质量、产出情况、变异检测、定量分析、物种、功能注释等多方面分析了NovaSeq X Plus的性能表现。
1) 产出高:单张FC产出3.6T
2) 测序质量高:平均Q30>96%
3) 变异检测精确度高:人全基因组测序文库和人全外显子测序文库SNP检测精确度可达99%。
4) 平台间一致性高:
01
各种文库类型产出及质量值表现
全基因组、全外显子、转录组、甲基化测序文库均具有较高产率,平均Q30>96%。
表1 各种文库类型产出及质量值
02
人全基因组测序文库性能表现
使用NA12878标准品进行建库,在NovaSeq X Plus和NovaSeq 6000平台同时进行人全基因组文库测序。
2.1 数据质控
与NovaSeq 6000相比,NovaSeq X Plus测序质量更高,平均Q30 94.36%,且在低GC和高GC区域覆盖度表现优异。
表2 人全基因组测序文库质控统计
注:hWGS_1为NovaSeq 6000测序文库,hWGS_1XL1和hWGS_1XL2为NovaSeq X Plus测序文库。
图1 NovaSeq X Plus GC偏好分布图
2.2 Mapping 统计
NovaSeq X Plus平台具有高度的数据均一性,在30x平均测序深度下,覆盖度达到99%。
表3 人全基因组测序文库mapping统计
2.3 变异检出
NovaSeq X Plus与NovaSeq 6000平台共有SNP可达到95%以上,且SNP和InDel精确度和灵敏度偏差<0.2%。
表4 人全基因组测序文库变异检测结果
图2 NovaSeq X Plus和NovaSeq 6000共有SNP维恩图
03
人全外显子测序文库性能表现
使用 Agilent SureSelect Human All Exon V6试剂盒,构建NA12878全外显子组文库,并在NovaSeq X Plus和NovaSeq 6000平台上同时进行测序。
3.1 数据质控
与NovaSeq 6000相比,NovaSeq X Plus Q30值更高,平均为97.17%。
表5 人全外显子测序文库质控统计
注:hWES_1为NovaSeq 6000测序文库,hWES_1XL1和hWES_1XL2为NovaSeq X Plus测序文库。
3.2 Mapping统计
NovaSeq X Plus 实现了对整个外显子区的均匀覆盖,覆盖率为99.5%,on target捕获效率为 67.05%。
表6 人全外显子文库mapping统计
图3 人全外显子文库捕获效率
3.3 变异检出
NovaSeq X Plus与NovaSeq 6000平台共有SNP可达到96%以上,且SNP和InDel精确度和灵敏度偏差<0.5%。
表7 人全外显子文库变异检测结果
图4 NovaSeq X Plus和NovaSeq 6000共有SNP维恩图
04
真核普通转录组文库性能表现
对人标准品UHRR和模式物种小鼠、拟南芥、线虫进行真核普通转录组文库构建,在NovaSeq X Plus和NovaSeq 6000平台同时进行真核普通转录组文库测序。
4.1 数据质控
相比NovaSeq 6000, NovaSeq X Plus质量值更高,平均Q30达到97.17%。
表8 真核普通转录组文库质控统计
注:后缀含X的样本为NovaSeq X Plus测序文库。
4.2 Mapping统计
Mapping率均达到95%以上,平台间偏差<1%。
表9 真核普通转录组文库mapping统计
4.3 定量分析
试测人标准品及多种模式生物(果蝇,拟南芥,线虫),平台间相关性均能达到0.97以上。
图5 NovaSeq X Plus和NovaSeq 6000中真核普通转录组文库相关性分析
注:左上为UHRR,左下为拟南芥,右上为果蝇,右下为线虫
05
宏基因组文库性能表现
5.1 数据质控及组装
相比NovaSeq 6000, NovaSeq X Plus质量值更高,平均Q30达到96.54%。组装指标N50, N90优于NovaSeq 6000。
表10 宏基因组文库质控及组装统计
注:后缀含X的样本为NovaSeq X Plus测序文库。
5.2 基因预测及丰度
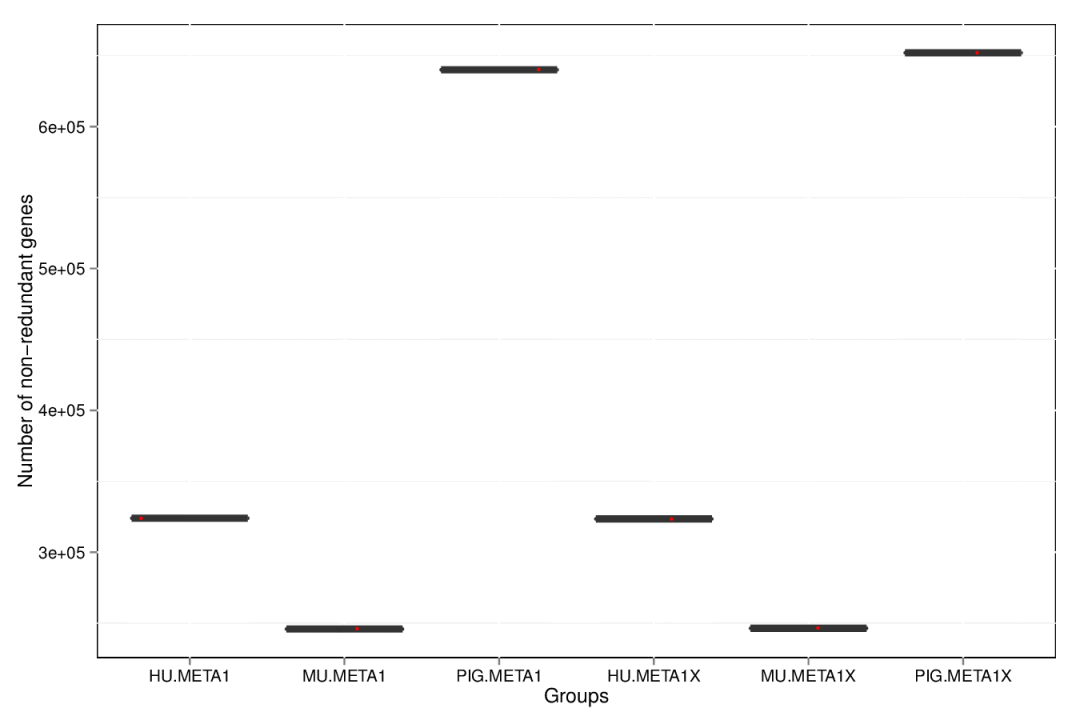
在<4*10^5基因数目水平下,平台间共有基因>94%,相关性>0.96。
图6 NovaSeq 6000和NovaSeq X Plus基因数目差异箱图
图7 NovaSeq 6000和NovaSeq X Plus相关系数热图
图8 NovaSeq 6000和NovaSeq X Plus基因数目韦恩图分析
5.3 物种、功能注释
平台间物种、功能注释(CAZy, eggNOG, KEGG, ARGs)具有高度一致性。
图9 基于Bray-Curtis距离的聚类树
注:从上至下依次为 物种门水平,CAZy ,eggNOG,KEGG的结果展示。图左侧是 Bray-Curtis 距离聚类树结构;右侧为各样品在门水平上的物种相对丰度分布和第一层级上的功能相对丰度分布
*备注(上下滑动查看)
(1) Effective: 过滤后用于后续的生物信息分析的Clean reads占原始数据的比例
(2) Error: 所有碱基的平均错误率
(3) Q20: Phred 数值大于20的碱基占总体碱基的百分比
(4) Q30: Phred 数值大于30的碱基占总体碱基的百分比
(5) GC(%):计算碱基G和C的数量总和占总的碱基数量的百分比
(6)Containing N:因单端测序Read中N含量超过该条Read碱基数10%而过滤掉的Reads对数及其占总Raw reads对数的比例
(7)Low quality:因单端测序Read中低质量(低于5)碱基比例超过Read长度的50%而过滤掉的Reads对数及其占总Raw reads对数的比例
(8)Adapter related:因有接头而过滤掉的Reads对数及其占总Raw reads对数的比例
(9) Mapped: 比对到参考基因组上的总reads数目 (比例)
(10) Properly mapped: 比对到参考基因组且插入片段大小正确的reads数目 (比例)
(11) PE mapped: 双端reads比对到参考基因组上的reads数目 (比例)
(12) Average sequencing depth: 比对到参考基因组的平均测序深度 (测序数据量/基因组大小)
(13) Coverage: 比对数据对全基因组区域的覆盖度 (碱基覆盖长度占全基因组碱基总长的比例)
(14) 4X: 全基因组区域中碱基覆盖深度不低于4X的比例
(15) 10X: 全基因组区域中碱基覆盖深度不低于10X的比例
(16) 20X: 全基因组区域中碱基覆盖深度不低于20X的比例
(17) Precision:精确率=TP/(TP+FP)(百分比)
(18) Recall:召回率=TP/(TP+FN)(百分比)
(19) F-score:F值=2 *精确率*召回率/(精确率+召回率)
(20) dbSNP (SNP): 在dbSNP数据库中报道的SNP数目与总SNP数目之比
(21) dbSNP (InDel): 在dbSNP数据库中报道的InDel数目与总InDel数目之比
(22) Unique_map:与参考基因组唯一位置对齐的reads数和百分比(用于后续定量数据分析),唯一映射率:(唯一映射的reads数)/(总reads数)*100。
(23) Multi_map:在参考基因组中与多个位置对齐的reads数和百分比,多重映射率:(多重映射reads)/(总reads)*100。
(24) N50 Len.(bp) : 表示将 Scaftigs 按照长度进行排序,然后由长到短加和,当加和值达到 Scaftigs 总长的 50% 时的 Scaftigs 的长度值
(25) N90 Len.(bp) : 表示将 Scaftigs 按照长度进行排序,然后由长到短加和,当加和值达到 Scaftigs 总长的 90%时的 Scaftigs 的长度值
全基因组、全外显子、转录组和宏基因组文库在NovaSeq X Plus平台均展现出更好的测序质量,并且与NovaSeq 6000具有高度一致性。其在数据质量、运行周期的出色表现,更有助于研究者深入加速挖掘群体基因组学、基因功能和调控相关机制。
故事未完,NovaSeq X Plus在非编码RNA和表观方向的性能表现请听下回分解。
诺禾致源专注于开拓前沿分子生物学技术和高性能计算在生命科学研究和人类健康领域的应用。作为世界上拥有最大测序能力之一的公司,我们利用深厚的科学知识、一流的客户服务和无与伦比的数据质量,帮助客户在基因组学这个不断发展的领域实现他们的研究目标。诺禾致源致力于成为您值得信赖的基因组合作伙伴。
客户服务中心热线:4006581585
邮箱:service@novogene.com
诺禾致源NovaSeq™ X Plus平台数据详解(上)
2023-05-04 04:37 星期四
诺禾致源






















要闻
股市
03-31 16:48 星期一
03-31 16:09 星期一
03-31 13:03 星期一
03-31 15:40 星期一
03-31 13:59 星期一
03-31 14:30 星期一
03-31 13:36 星期一
03-31 13:37 星期一
03-31 13:17 星期一
03-31 12:53 星期一










财联社 ©2018-2025上海界面财联社科技股份有限公司 版权所有沪ICP备14040942号-9沪公网安备31010402006047号互联网新闻信息服务许可证:31120170007沪金信备 [2021] 2号